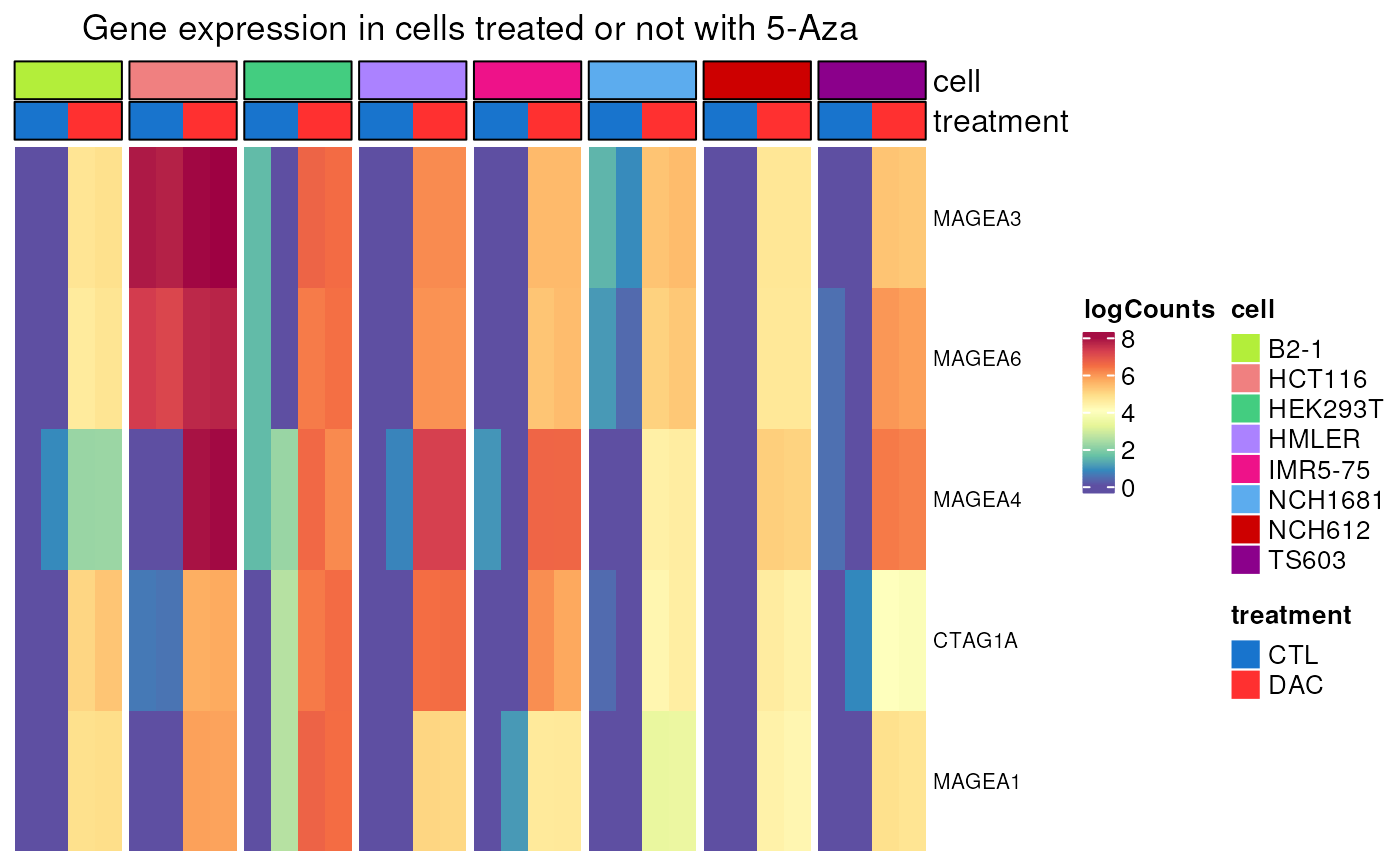
Gene expression in cells treated or not by a demethylating agent
Source:R/DAC_induction.R
DAC_induction.RdPlots a heatmap of normalised gene counts (log-transformed) in a selection of cells treated or not by 5-Aza-2'-Deoxycytidine (DAC), a demethylating agent.
Arguments
- genes
characternaming the selected genes. The default value,NULL, takes all CT specific genes.- multimapping
logical(1)defining whether to use multi-mapped gene expression datasetCTdata::DAC_treated_cells_multimappingorDAC_treated_cells. Default isTRUE.- include_CTP
logical(1)IfTRUE, CTP genes are included. (FALSEby default).- values_only
logical(1). IfTRUE, the function will return the gene normalised logcounts in all samples instead of the heatmap. Default isFALSE.
Value
A heatmap of selected genes in cells treated or not by a
demethylating agent. If values_only is TRUE, gene normalised
logcounts are returned instead.
Details
RNAseq data from cells treated or not with 5-aza downloaded from
SRA. (SRA references and information about cell lines and DAC
treatment are stored the colData of DAC_treated_cells). Data was
processed using a standard RNAseq pipeline.
hisat2 was used
to align reads to grch38 genome.
featurecounts
was used to assign reads to genes. Note that -M parameter was used
or not to allow or not counting multi-mapping reads.
Examples
DAC_induction(genes = c("MAGEA1", "MAGEA3", "MAGEA4", "MAGEA6", "CTAG1A"))
#> see ?CTdata and browseVignettes('CTdata') for documentation
#> loading from cache
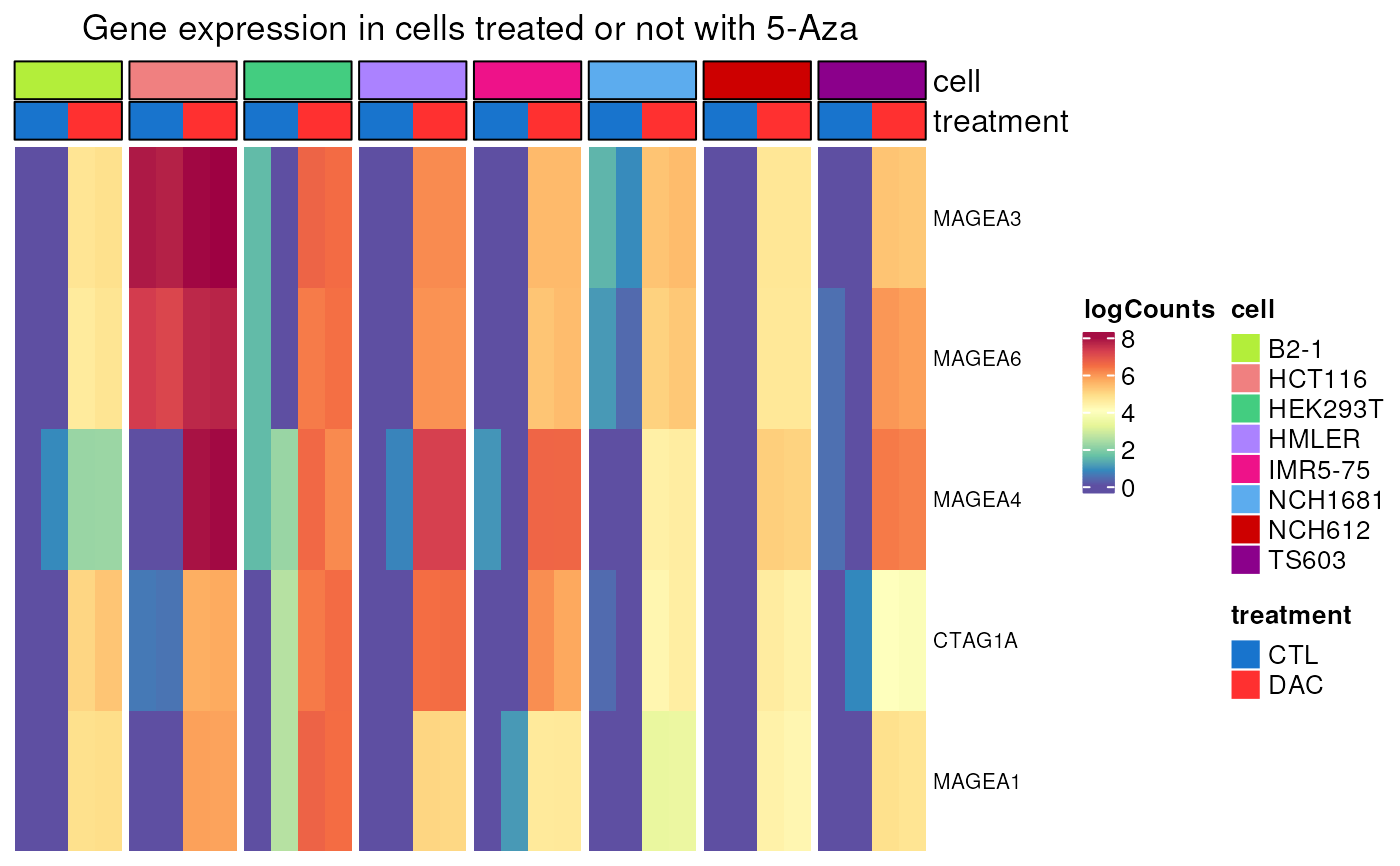 DAC_induction(genes = c("MAGEA1", "MAGEA3", "MAGEA4", "MAGEA6", "CTAG1A",
multimapping = FALSE))
#> see ?CTdata and browseVignettes('CTdata') for documentation
#> loading from cache
#> Warning: 1 out of 6 names invalid: FALSE.
#> See the manual page for valid types.
DAC_induction(genes = c("MAGEA1", "MAGEA3", "MAGEA4", "MAGEA6", "CTAG1A",
multimapping = FALSE))
#> see ?CTdata and browseVignettes('CTdata') for documentation
#> loading from cache
#> Warning: 1 out of 6 names invalid: FALSE.
#> See the manual page for valid types.