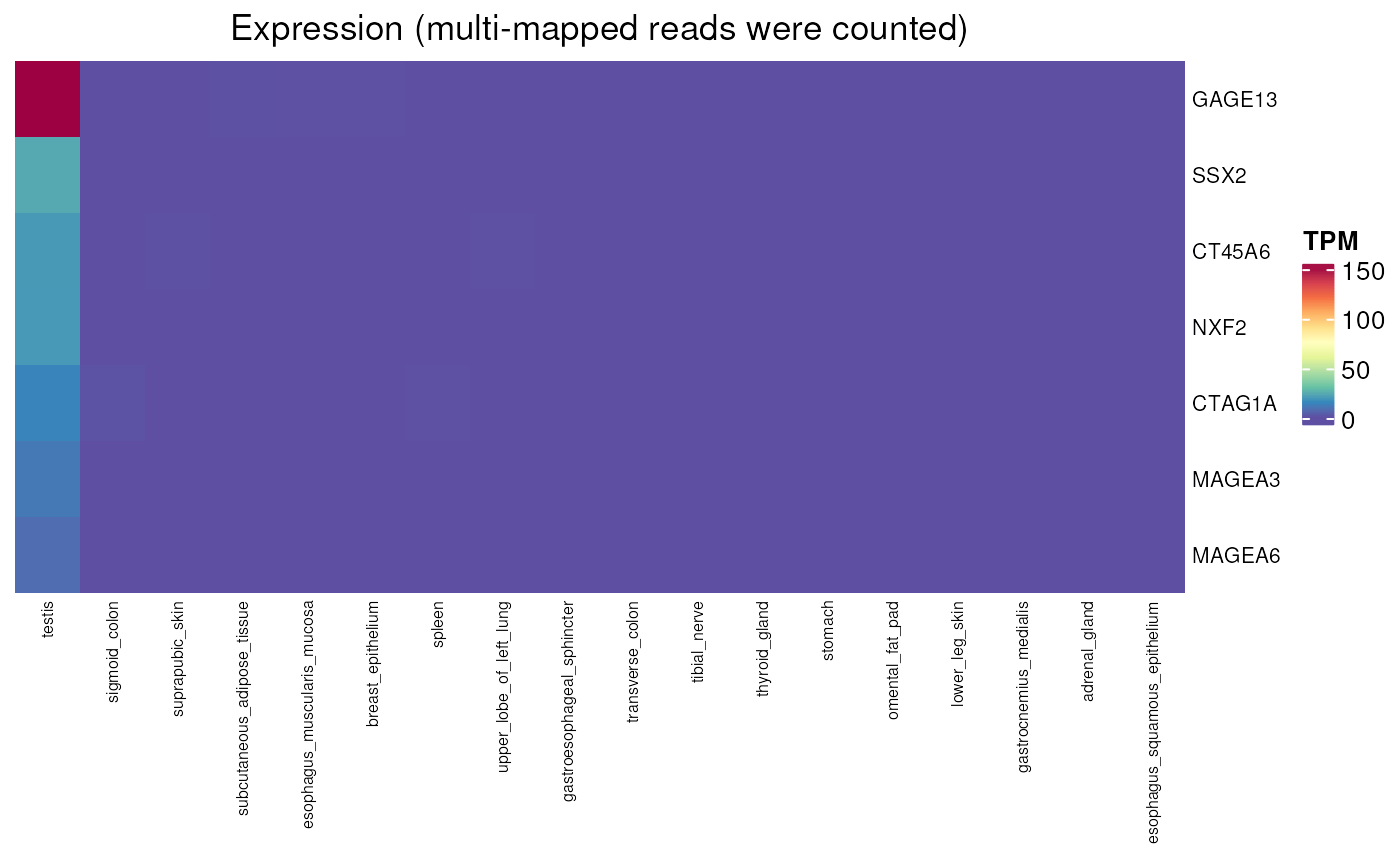
Expression values (TPM) of genes in normal tissues with or without multimapping
Source:R/normal_tissue_expression_multimapping.R
normal_tissue_expression_multimapping.RdPlots a heatmap of gene expression values in a set of normal tissues. Expression values (in TPM) have been evaluated by either counting or discarding multi-mapped reads. Indeed, many CT genes belong to gene families from which members have identical or nearly identical sequences. Some CT can only be detected in RNAseq data in which multimapping reads are not discarded.
Usage
normal_tissue_expression_multimapping(
genes = NULL,
include_CTP = FALSE,
multimapping = TRUE,
units = c("TPM", "log_TPM"),
values_only = FALSE
)Arguments
- genes
characternaming the selected genes. The default value,NULL, takes all CT (specific) genes.- include_CTP
logical(1)IfTRUE, CTP genes are included. (FALSEby default).- multimapping
logical(1)that specifies if returned expression values must take into account or not multi-mapped reads. TRUE by default.- units
character(1)with expression values unit. Can be"TPM"(default) or"log_TPM"(log(TPM + 1)).- values_only
logical(1). IfTRUE, the function will return the expression values in all samples instead of the heatmap. Default isFALSE.
Value
A heatmap of selected gene expression values in a set of
normal tissues calculated by counting or discarding
multi-mapped reads. If values_only = TRUE, gene expression values
are returned instead.
Details
RNAseq data from a set of normal tissues were downloaded from Encode. (see inst/scripts/make_CT_normal_tissues_multimapping.R for fastq references) Fastq files were processed using a standard RNAseq pipeline including FastQC for the quality control of the raw data, and trimmomatic to remove low quality reads and trim the adapter from the sequences. hisat2 was used to align reads to grch38 genome. featurecounts was used to assign reads to genes using Homo_sapiens.GRCh38.105.gtf.
Two different pipelines were run in order to remove or not multi-mapping reads. When multimapping was allowed, hisat2 was run with -k 20 parameter (reports up to 20 alignments per read), and featurecounts was run with -M parameter (multi-mapping reads are counted).
Examples
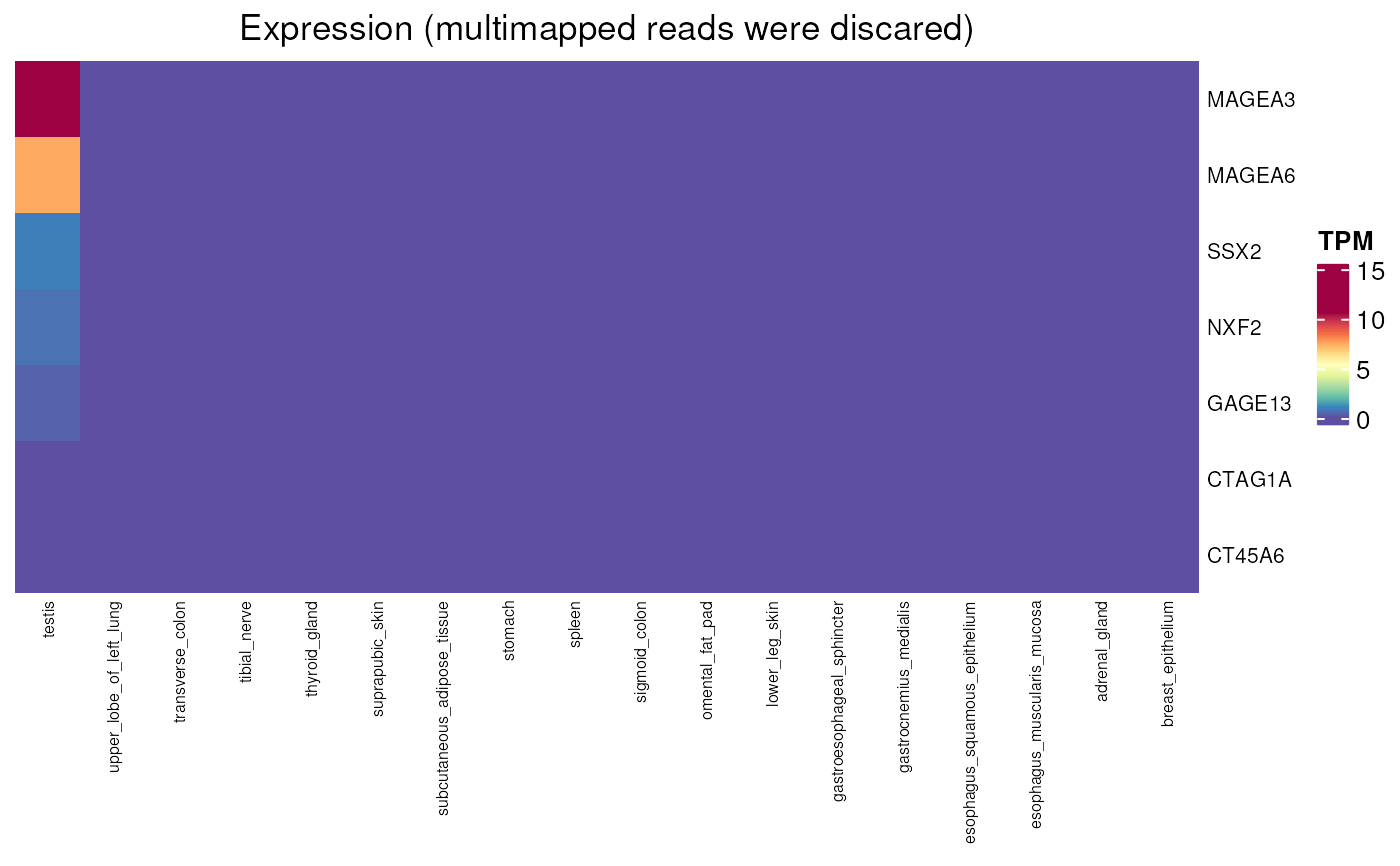
normal_tissue_expression_multimapping(
genes = c("GAGE13", "CT45A6", "NXF2", "SSX2", "CTAG1A",
"MAGEA3", "MAGEA6"), multimapping = FALSE)
#> see ?CTdata and browseVignettes('CTdata') for documentation
#> loading from cache
 normal_tissue_expression_multimapping(
genes = c("GAGE13", "CT45A6", "NXF2", "SSX2", "CTAG1A",
"MAGEA3", "MAGEA6"), multimapping = TRUE)
#> see ?CTdata and browseVignettes('CTdata') for documentation
#> loading from cache
normal_tissue_expression_multimapping(
genes = c("GAGE13", "CT45A6", "NXF2", "SSX2", "CTAG1A",
"MAGEA3", "MAGEA6"), multimapping = TRUE)
#> see ?CTdata and browseVignettes('CTdata') for documentation
#> loading from cache