Promoter methylation of any gene in normal tissues
Source:R/normal_tissues_mean_methylation.R
normal_tissues_mean_methylation.RdPlots a heatmap of mean promoter methylation levels of any genes in normal tissues. Methylation levels in tissues correspond to the mean methylation of CpGs located in range of 1000 pb upstream and 200 pb downstream from gene TSS.
Usage
normal_tissues_mean_methylation(
genes = NULL,
include_CTP = FALSE,
values_only = FALSE,
na.omit = TRUE
)Arguments
- genes
characternaming the selected genes. The default value,NULL, takes all CT (specific) genes.- include_CTP
logical(1)IfTRUE, CTP genes are included. (FALSEby default).- values_only
logical(1),FALSEby default. IfTRUE, the function will return the methylation values in all samples instead of the heatmap.- na.omit
logical(1)specifying if genes with missing methylation values in some tissues should be removed (TRUEby default). Note that no gene clustering will be done when methylation values are missing.
Value
Heatmap of mean promoter methylation of any
gene in normal tissues. If values_only = TRUE, methylation values
are returned instead.
Examples
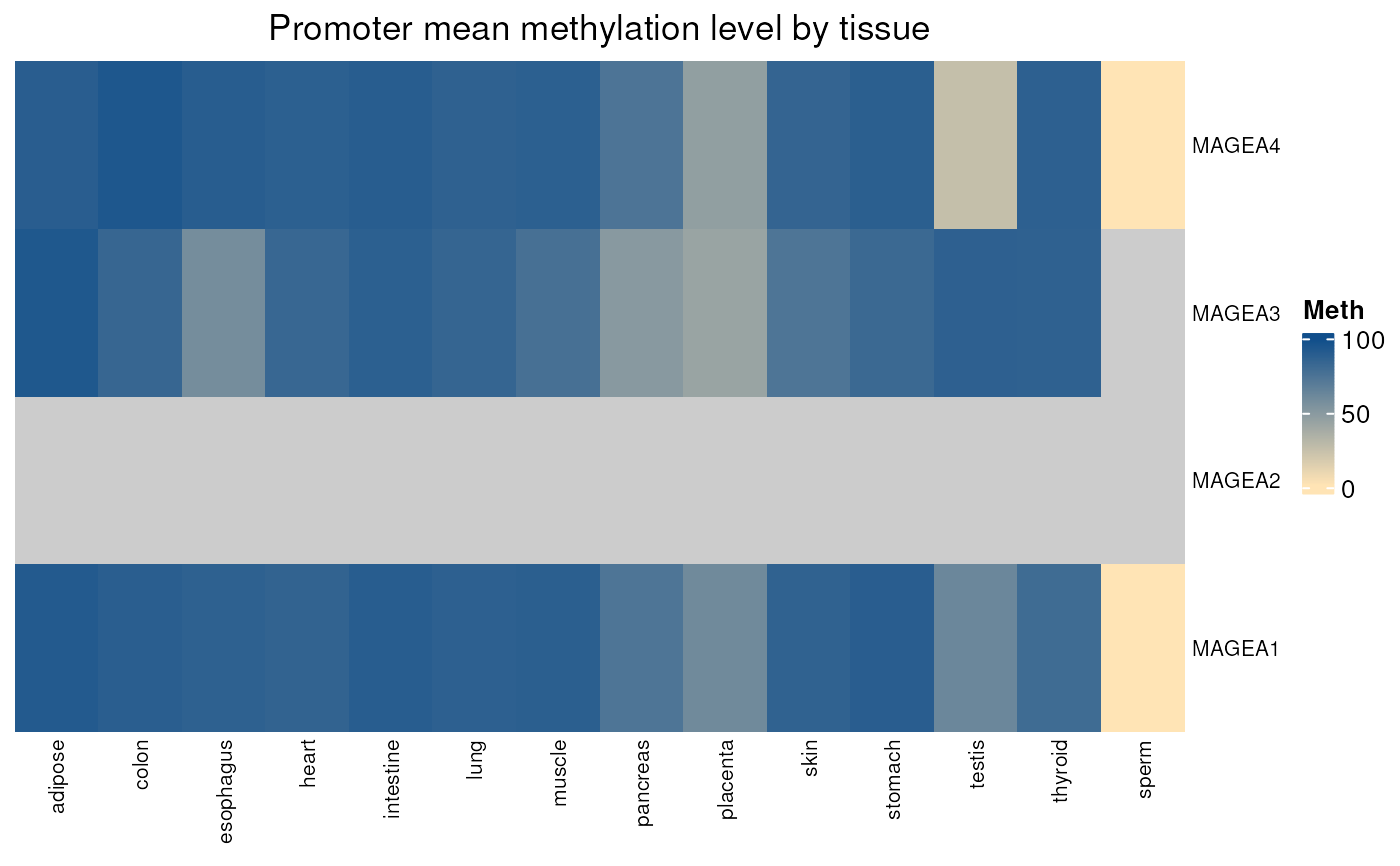
normal_tissues_mean_methylation()
#> see ?CTdata and browseVignettes('CTdata') for documentation
#> loading from cache
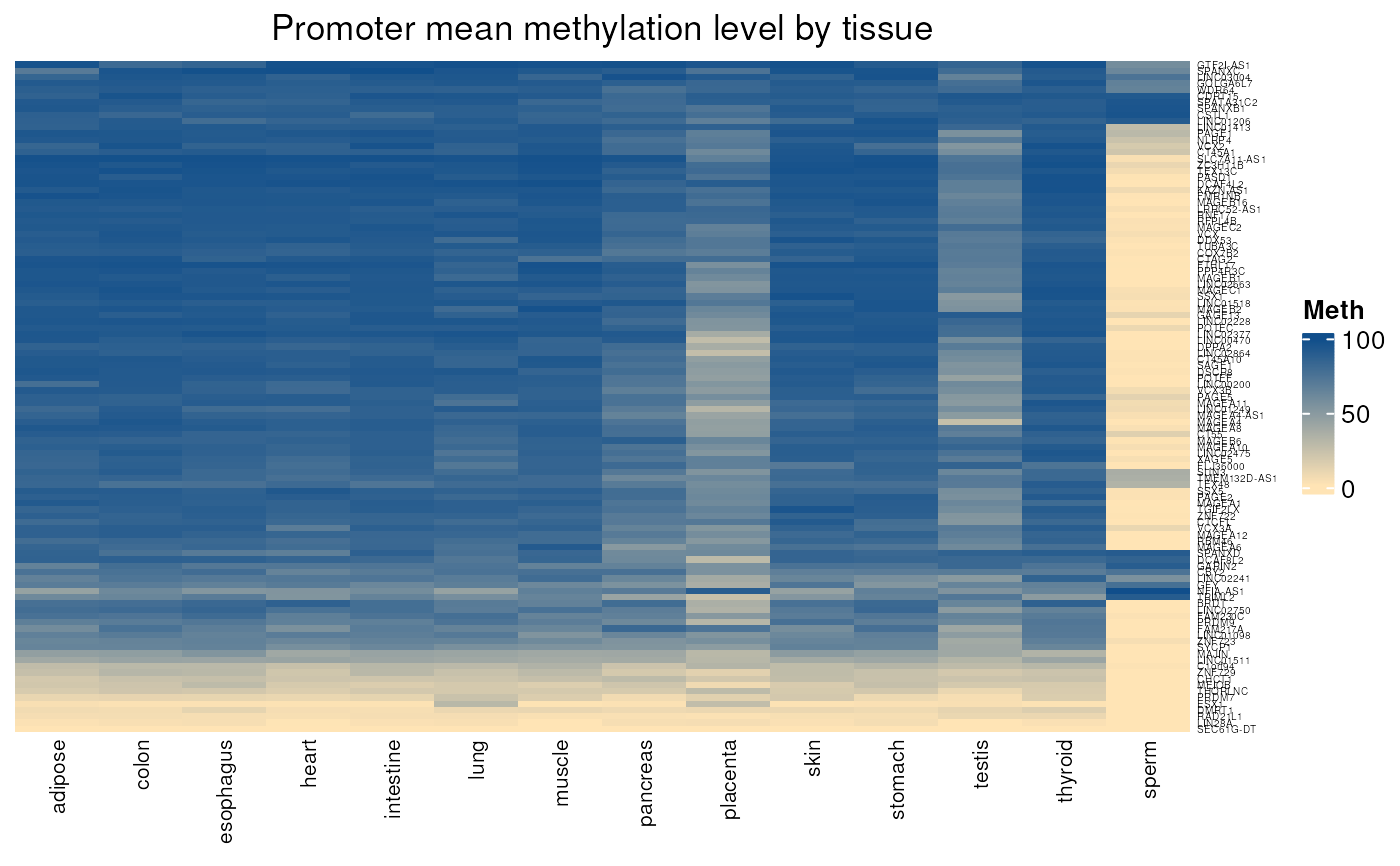 normal_tissues_mean_methylation(c("MAGEA1", "MAGEA2", "MAGEA3", "MAGEA4"))
#> see ?CTdata and browseVignettes('CTdata') for documentation
#> loading from cache
normal_tissues_mean_methylation(c("MAGEA1", "MAGEA2", "MAGEA3", "MAGEA4"))
#> see ?CTdata and browseVignettes('CTdata') for documentation
#> loading from cache
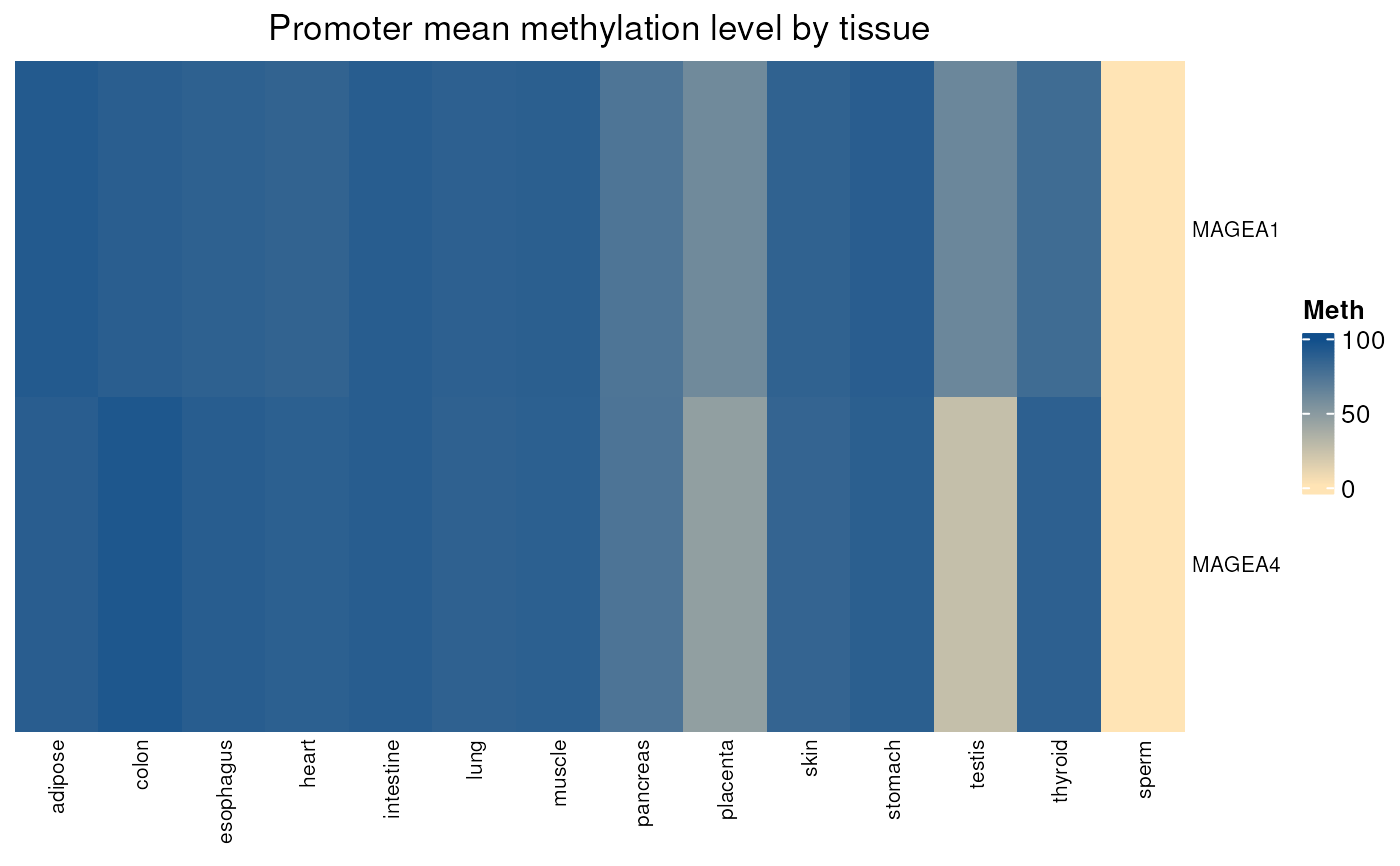 normal_tissues_mean_methylation(c("MAGEA1", "MAGEA2", "MAGEA3", "MAGEA4"),
na.omit = FALSE)
#> see ?CTdata and browseVignettes('CTdata') for documentation
#> loading from cache
normal_tissues_mean_methylation(c("MAGEA1", "MAGEA2", "MAGEA3", "MAGEA4"),
na.omit = FALSE)
#> see ?CTdata and browseVignettes('CTdata') for documentation
#> loading from cache