Chapter 4 Manipulating sequences with Biostrings
Learning objectives
In this chapter, we will learn about a new object, that is specialised for the manipulation of biological sequences.
- Learn about the fasta format format for biological sequences.
- Learn about the
*StringSetclasses. - Working with whole genomes.
- Find patterns in sequence data.
This chapter is based on the course material presented by Martin Morgan during the Bioinformatics Summer School 2019 at Louvain-la-Neuve, Belgium.
4.1 Working with sequences - a first start
It is easy to use standard R character vectors to define DNA sequences. Below, we create a vector with three short sequences.
## [1] "AAATCGA" "ATACAACAT" "TTGCCA"We can ask about properties of these sequences and perform some operations using base R function:
## [1] 3## [1] 7 9 6## [1] "AAATCGA" "TTGCCA"## [1] "TTGCCA" "AAATCGA" "ATACAACAT"However, once we want to perform biologically relevant operations, base R fails us; it has no notion of operations relevant to DNA sequences, e.g.,
Likewise, we can name a variable anything, the semantic meaning of the variable name is not enforced by R
4.2 Working with sequences - using Biostrings
The Bioconductor Biostrings package can do all that base R can do, in addition to knowing about the semantics of the sequences is handles. Let’s start by loading the package:
For more information about the Biostrings package, see the vignettes
available online the package
package.
4.2.1 DNAStringSet()
The DNAStringSet class that knows about DNA sequences. We can
easily create a DNAStringSet from our character vector with
## DNAStringSet object of length 3:
## width seq
## [1] 7 AAATCGA
## [2] 9 ATACAACAT
## [3] 6 TTGCCA► Question
Does the object dna support the operations illustrated above for a
character vector, especially length(), nchar(), [, and
sample()?
► Solution
► Question
Prove to yourself that at least some other useful, DNA-specific,
functions exist, e.g., reverse() and reverseComplement().
► Solution
► Question
What happens when you try to create a DNAStringSet() from an object
such as not_a_dna_sequence, defined above, that does not contain a
DNA sequence? Warning: the message is quite cryptic, can you provide a
‘human’ translation?
► Solution
► Question
Why does DNAStringSet("ACGTMRW") not create an error, since MRW
are not standard nucleotides? For hints, see the section ’The DNA
alphabet:” in the help page ?DNAString and the ?IUPAC_CODE_MAP
manual.
► Question
What is the difference between a DNAString and a DNAStringSet?
► Solution
4.2.2 Learning more about a class
The function DNAStringSet() returns an object that has a
particular class
## [1] "DNAStringSet"
## attr(,"package")
## [1] "Biostrings"Associated with the class are a series of methods (i.e. functions that have a behaviour that is tuned for that class) that operate on the class.
► Question
Discover many (unfortunately, not all) methods acting on
an object of class DNAStringSet using methods(class = "DNAStringSet"). Verify that reverseComplement is among those
methods.
Help pages describing a particular method can be found using ?, with
the search query quoted and with tab-completion providing hints on
what the appropriate help topic is.
► Question
Find the help page for the reverseComplement method operating on a
DNAStringSet object, using ?reverseComplement.
Help pages provide a description of the technical details required for creating classes and using methods. Vignettes provide a more narrative description of overall package use.
► Question
Use browseVignettes(package = "Biostrings") to see vignettes
available for this package; explore a few vignettes to get a sense of
possible content.
4.3 Reading DNA sequence data from a file
It is unlikely that we would enter 1000’s of DNA sequences ‘by
hand’. Instead, we might read the data from a standard file
format. For DNA sequences the standard file format is often a ‘FASTA’
file, sometimes abbreviated with an extension .fa and often
compressed with an additional extension .fa.gz. An example of a
FASTA file containing DNA sequences of the 2000bp upstream nucleotides
of all genes annotated in the Drosophila melanogaster dm3 genome
build, is distributed with the Biostrings package. Here’s the path
to the FASTA file.
► Question
Take a peak at the structure of a FASTA file by looking at the first
five lines. You can use the readLines function, setting the n
parameter to read only a limited number of files (see readLines).
► Solution
In this case, the first line is an identifier, containing information
about the gene NM_078863 as well as the genomic coordinates of the
sequence chr2L:16764737-16766736. The next lines are the DNA
sequence. After a certain number of lines, a new record starts.
## [1] "cacgcacaccgatcgtcgaatcgaaaagctttcggggtcttacgggatcc"
## [2] "atgggtatcaagttgccccgtataaaaggcaagtttaccggttgcacggt"
## [3] ">NM_001201794_up_2000_chr2L_8382455_f chr2L:8382455-8384454"
## [4] "ttatttatgtaggcgcccgttcccgcagccaaagcactcagaattccggg"
## [5] "cgtgtagcgcaacgaccatctacaaggcaatattttgatcgcttgttagg"We could fairly easily write our own parser for this format, but
this would be error-prone and unnecessary. Instread, we want to use
the readDNAStringSet function from the Biostrings package for
that.
## DNAStringSet object of length 26454:
## width seq names
## [1] 2000 GTTGGTGGCCCACCAGTGCCA...CAAGTTTACCGGTTGCACGGT NM_078863_up_2000...
## [2] 2000 TTATTTATGTAGGCGCCCGTT...ATACGGAAAGTCATCCTCGAT NM_001201794_up_2...
## [3] 2000 TTATTTATGTAGGCGCCCGTT...ATACGGAAAGTCATCCTCGAT NM_001201795_up_2...
## [4] 2000 TTATTTATGTAGGCGCCCGTT...ATACGGAAAGTCATCCTCGAT NM_001201796_up_2...
## [5] 2000 TTATTTATGTAGGCGCCCGTT...ATACGGAAAGTCATCCTCGAT NM_001201797_up_2...
## ... ... ...
## [26450] 2000 ATTTACAAGACTAATAAAGAT...AAAATTAAATTTCAATAAAAC NM_001111010_up_2...
## [26451] 2000 GATATACGAAGGACGACCTGC...TTGTTTGAGTTGTTATATATT NM_001015258_up_2...
## [26452] 2000 GATATACGAAGGACGACCTGC...TTGTTTGAGTTGTTATATATT NM_001110997_up_2...
## [26453] 2000 GATATACGAAGGACGACCTGC...TTGTTTGAGTTGTTATATATT NM_001276245_up_2...
## [26454] 2000 CGTATGTATTAGTTAACTCTG...TCAAAGTGTAAGAACAAATTG NM_001015497_up_2...► Question
Query the object for basic properties, e.g., it’s length() and that
number of character in each sequence (try table(nchar(dna))).
► Question
Use letterFrequency() to determine GC content of each of the DNA
sequences in dna. The letters argument should be "GC"; as.prob = TRUE returns values between 0 and 1. The data is returned as a
matrix with 1 column.
► Solution
► Question
What are the mean and standard deviation of the GC contents in the 26454 genes? What genes(s) has/have the highes GC content?
► Solution
► Question
Plot the distribution of GC frequencies in the dna object using base
graphics hist() and plot(density()), and using ggplot().
► Solution
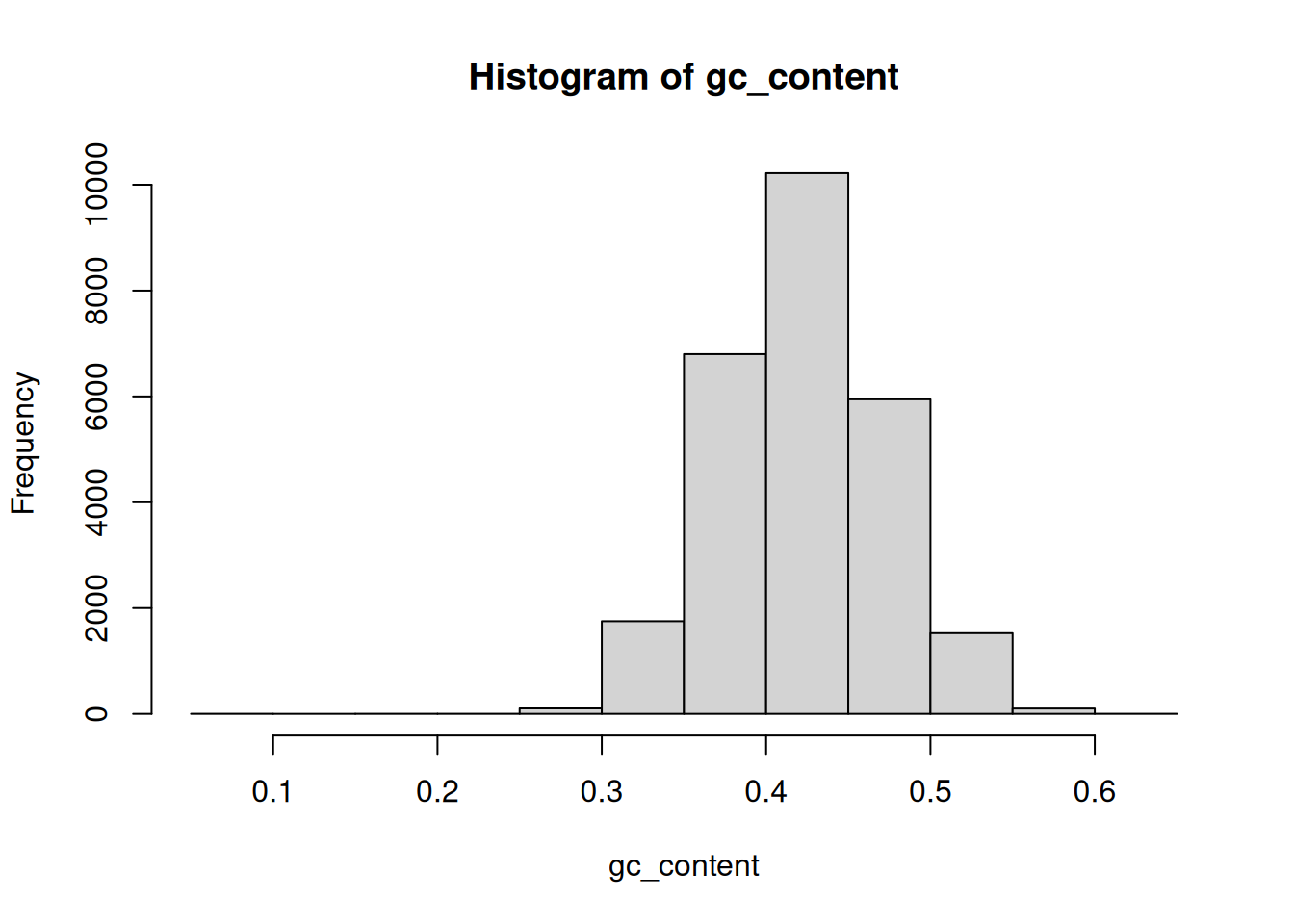
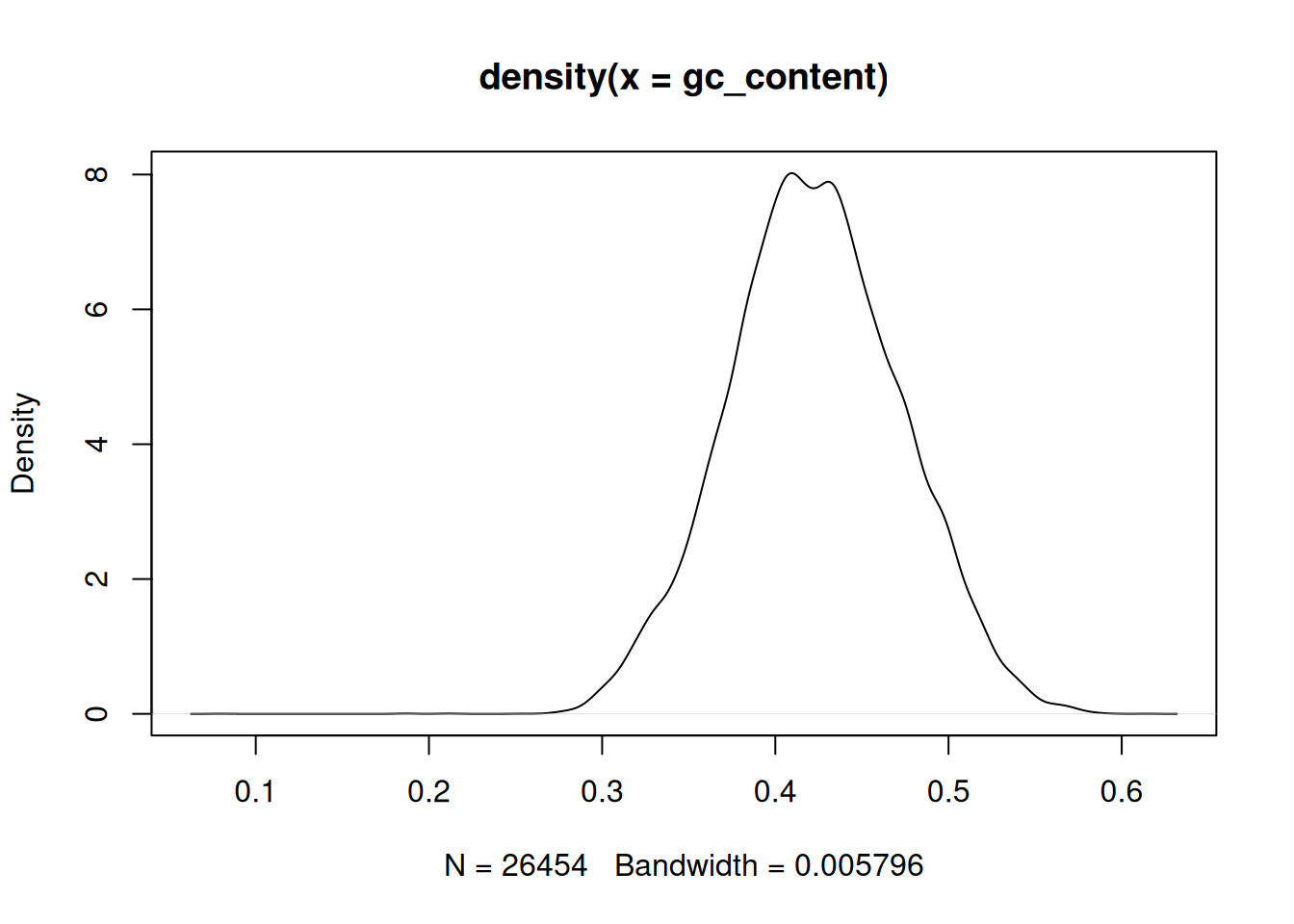
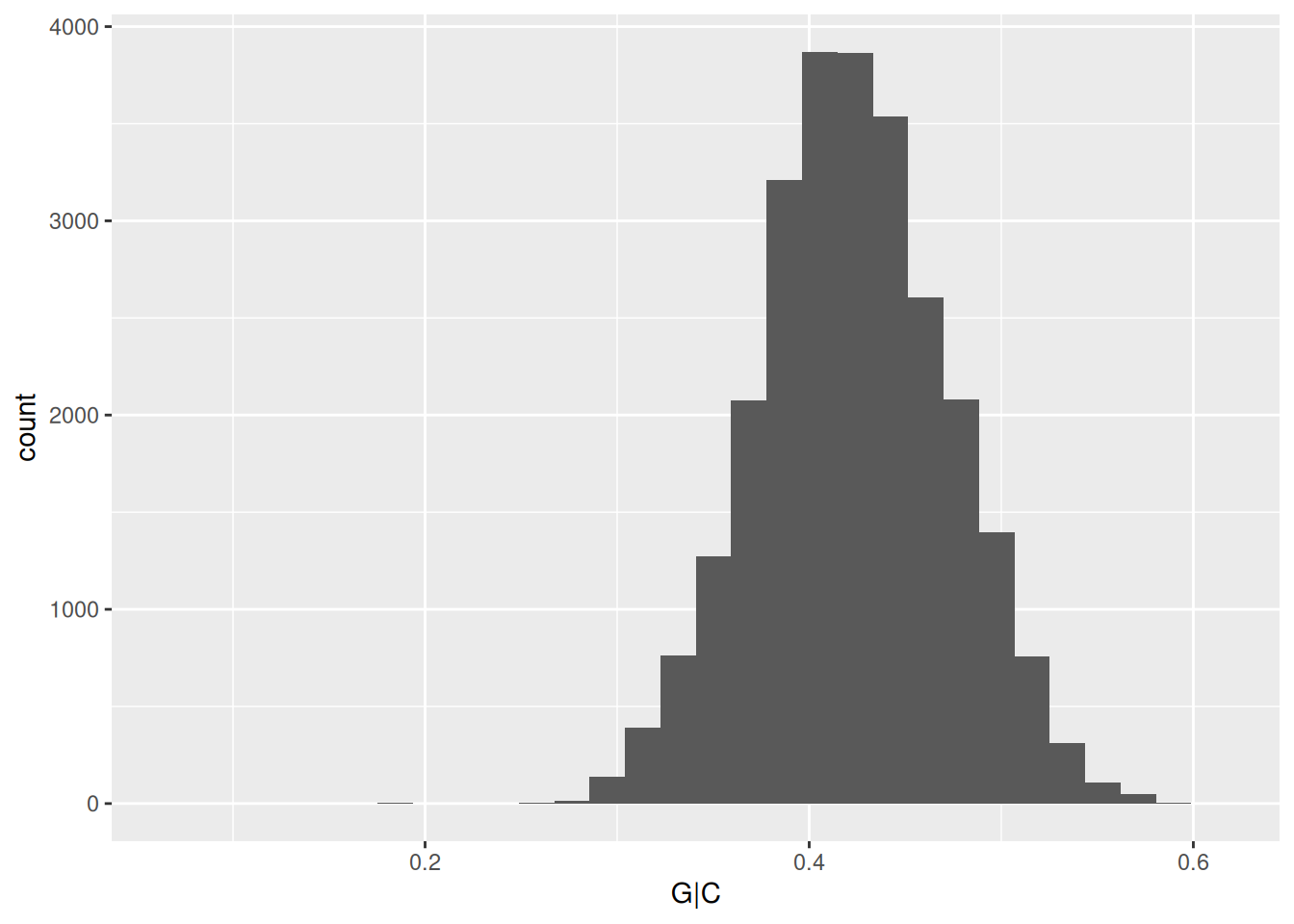
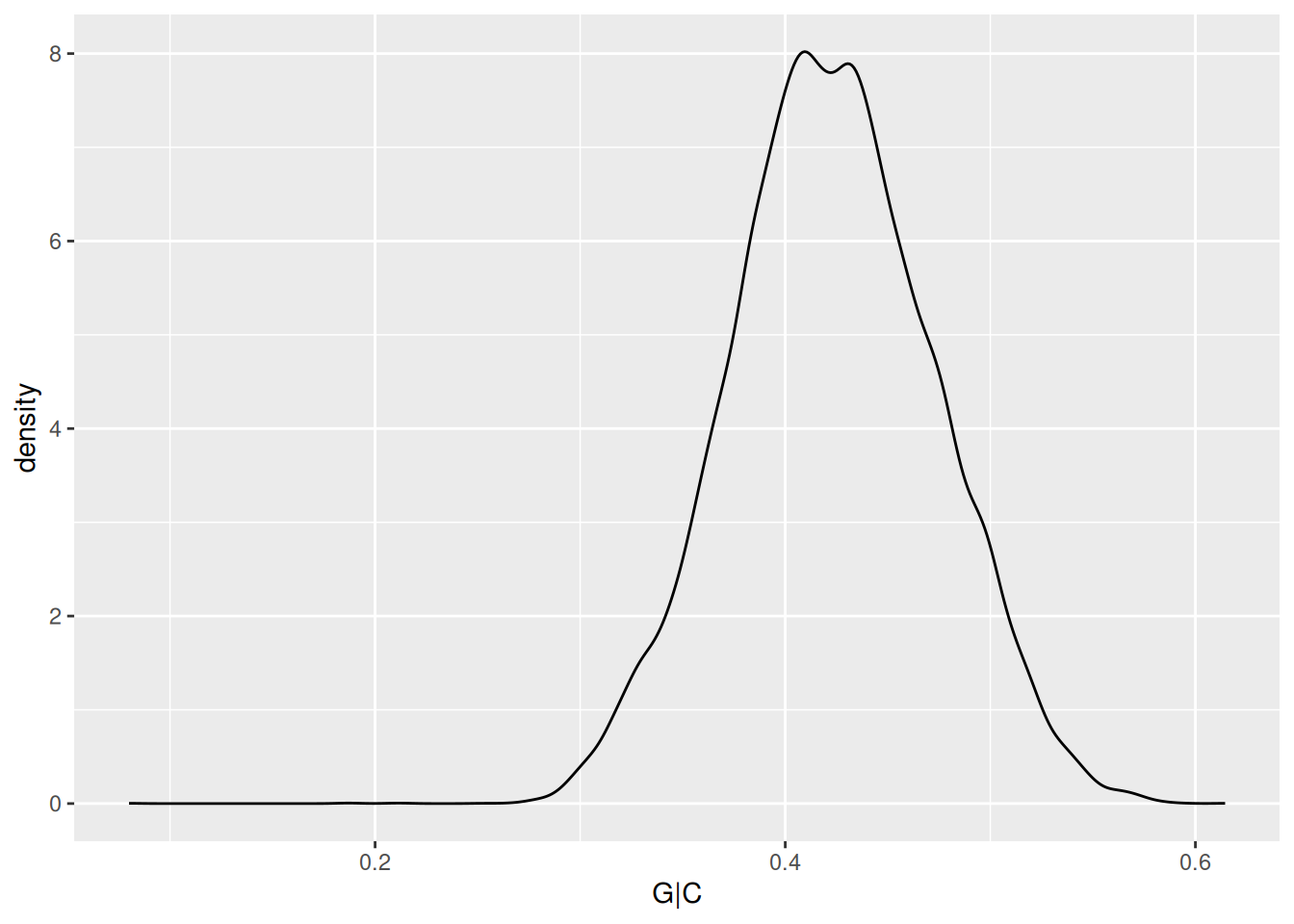
4.4 Tidy Bioconductor
Although Bioconductor emphasizes formal objects like DNAStringSet
rather than tibble-like data frames, some of the ways one interacts with tidy data can be
applied to Bioconductor objects. For instance, the GC content example
might be written in ‘traditional’ form as
but could be written using pipes and to reesult in a tibble for easier down-stream manipulation
## # A tibble: 26,454 × 1
## `G|C`
## <dbl>
## 1 0.378
## 2 0.43
## 3 0.43
## 4 0.43
## 5 0.43
## 6 0.43
## 7 0.43
## 8 0.43
## 9 0.43
## 10 0.43
## # ℹ 26,444 more rows4.5 Subsetting sequences
As [ is used to subset a DNAStringSet, it can’t be used to take
substrings of a sequence. This can be done with the subseq sequence.
## DNAStringSet object of length 26454:
## width seq names
## [1] 100 GTTGGTGGCCCACCAGTGCCA...CAATGACTCAAAACGAAAATG NM_078863_up_2000...
## [2] 100 TTATTTATGTAGGCGCCCGTT...ATATTTTGATCGCTTGTTAGG NM_001201794_up_2...
## [3] 100 TTATTTATGTAGGCGCCCGTT...ATATTTTGATCGCTTGTTAGG NM_001201795_up_2...
## [4] 100 TTATTTATGTAGGCGCCCGTT...ATATTTTGATCGCTTGTTAGG NM_001201796_up_2...
## [5] 100 TTATTTATGTAGGCGCCCGTT...ATATTTTGATCGCTTGTTAGG NM_001201797_up_2...
## ... ... ...
## [26450] 100 ATTTACAAGACTAATAAAGAT...GTGGGCGTTGCAACATGGATC NM_001111010_up_2...
## [26451] 100 GATATACGAAGGACGACCTGC...ATGACAGTCTTCTGGTGCTGG NM_001015258_up_2...
## [26452] 100 GATATACGAAGGACGACCTGC...ATGACAGTCTTCTGGTGCTGG NM_001110997_up_2...
## [26453] 100 GATATACGAAGGACGACCTGC...ATGACAGTCTTCTGGTGCTGG NM_001276245_up_2...
## [26454] 100 CGTATGTATTAGTTAACTCTG...TTACTGTAGTTAGGAAGATTA NM_001015497_up_2...## DNAStringSet object of length 26454:
## width seq names
## [1] 100 GTTGGTGGCCCACCAGTGCCA...CAATGACTCAAAACGAAAATG NM_078863_up_2000...
## [2] 100 TTATTTATGTAGGCGCCCGTT...ATATTTTGATCGCTTGTTAGG NM_001201794_up_2...
## [3] 100 TTATTTATGTAGGCGCCCGTT...ATATTTTGATCGCTTGTTAGG NM_001201795_up_2...
## [4] 100 TTATTTATGTAGGCGCCCGTT...ATATTTTGATCGCTTGTTAGG NM_001201796_up_2...
## [5] 100 TTATTTATGTAGGCGCCCGTT...ATATTTTGATCGCTTGTTAGG NM_001201797_up_2...
## ... ... ...
## [26450] 100 ATTTACAAGACTAATAAAGAT...GTGGGCGTTGCAACATGGATC NM_001111010_up_2...
## [26451] 100 GATATACGAAGGACGACCTGC...ATGACAGTCTTCTGGTGCTGG NM_001015258_up_2...
## [26452] 100 GATATACGAAGGACGACCTGC...ATGACAGTCTTCTGGTGCTGG NM_001110997_up_2...
## [26453] 100 GATATACGAAGGACGACCTGC...ATGACAGTCTTCTGGTGCTGG NM_001276245_up_2...
## [26454] 100 CGTATGTATTAGTTAACTCTG...TTACTGTAGTTAGGAAGATTA NM_001015497_up_2...## 10-letter DNAString object
## seq: GTTGGTGGCC## 10-letter DNAString object
## seq: GTTGGTGGCC► Question
The start, end and width arguments can be vector of length > 1
themselves so as to generate sequences of different lengths. Create a
new object that contains subsets of the first sequence of dna with,
respectively nucleotides 1 to 1, 1 to 2, 1 to 3, …, 1 to 10.
► Solution
4.6 Working with genomes
BSgenome packages contain whole genome sequences as distributed by
ENSEMBL, NCBI and others. In this next example we will load the whole
genome sequence for Drosophila melanogaster from UCSC’s dm2 build, and
calculate the GC content across chromosome 2L.
We first define the genomic range corresponding to chromosome 2L:
library("Biostrings")
library("BSgenome.Dmelanogaster.UCSC.dm2")
chr2L_range <- GRanges("chr2L", IRanges(1, seqlengths(Dmelanogaster)["chr2L"]))
chr2L_range## GRanges object with 1 range and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr2L 1-22407834 *
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengthsWe can now use the getSeq() fonction to extract the actual sequence
for the genome as a DNAStringSet object. We can use it as is to get
the whole genome …
## DNAStringSet object of length 13:
## width seq names
## [1] 22407834 CGACAATGCACGACAGAGGAA...TTAGTTGAAGGTGAGGGTGCT chr2L
## [2] 20766785 CAGGTACAGTATTCAGTACAG...CTGTTTGCATTCTAGGAATTC chr2R
## [3] 23771897 TAGGGAGAAATATGATCGCGT...AAAGAGAAGCCTATACTTGGG chr3L
## [4] 27905053 GAATTCTCTCTTGTTGTAGTC...CTGTTCGCATTCTAGGAATTC chr3R
## [5] 1281640 GAATTCGCGTCCGCTTACCCA...ATGTAGTATTTTTGTAAGCTT chr4
## ... ... ...
## [9] 1694122 ATTTTACGATACTGCTTATTT...CTAGGAAATTGGCAAAAACTC chr2h
## [10] 2955737 ACCTCAGGAGTCCGAAGTATG...AGATATCTCCATGATACTGAG chr3h
## [11] 88110 ACAAGGCAAGGCAAGGCAAGG...AGTTCGTCAAATGATTAAAAA chr4h
## [12] 359526 ATGCCAAAAAATTGGAATTTC...AGATAGATAGATAGATAGATA chrXh
## [13] 396896 CCAAAGGTTATCCAATCCGGA...ACTATAATAGACTTTATCCTA chrYh… or extract the region of interest with the GRranges object
previously defined:
## DNAStringSet object of length 1:
## width seq
## [1] 22407834 CGACAATGCACGACAGAGGAAGCAGAACAGAT...AAAGTTTCGAGTTAGTTGAAGGTGAGGGTGCT## G|C
## [1,] 0.4190165► Question
Using letterFrequency, calculate the number and frequency of A, G, C
and T nucleotides in chromosome 2L.
► Solution
4.7 Additional exercises
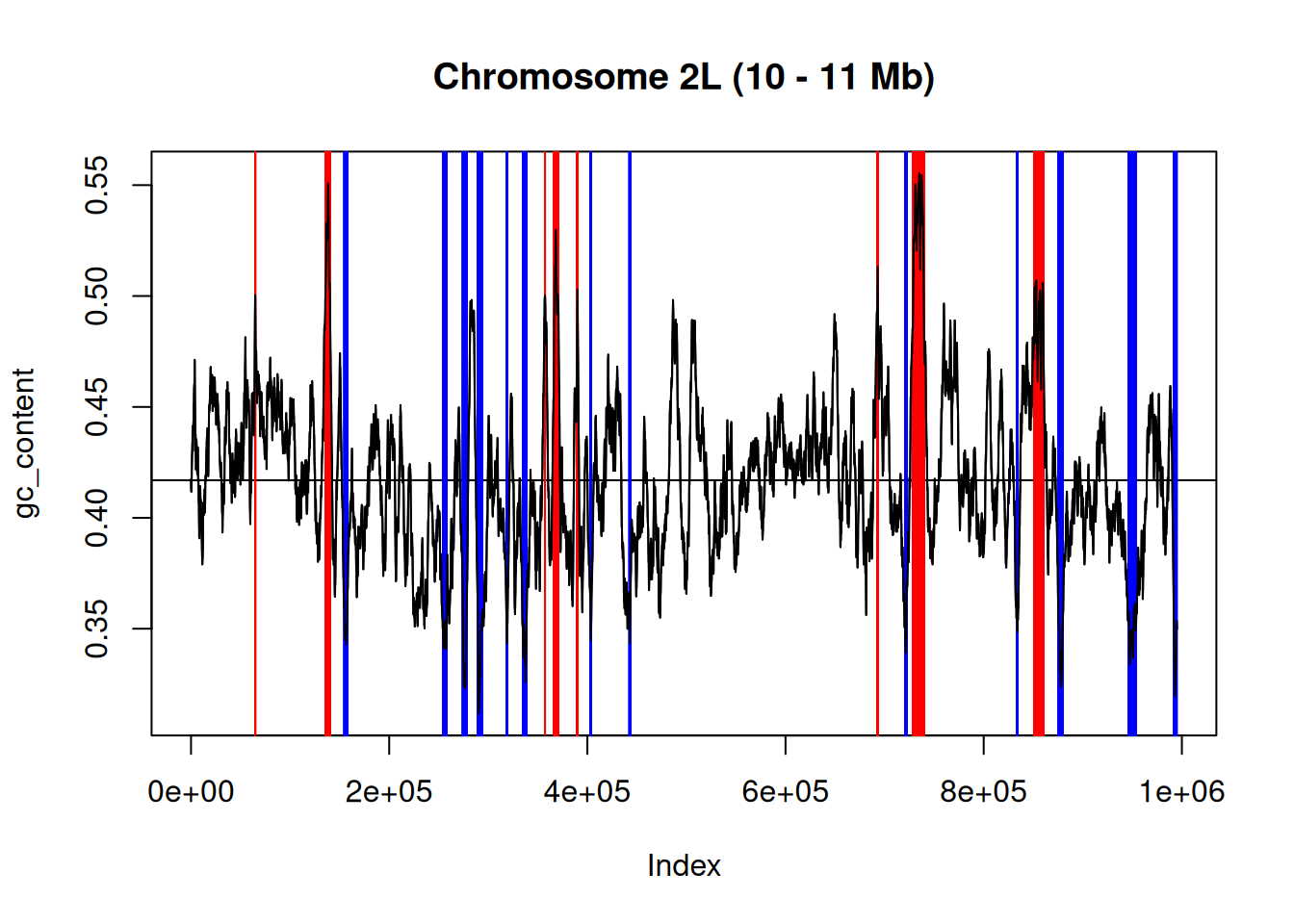
► Question
Focusing on the region from base 10 million to 11 million along
chromosome 2L, calculate the GC content across a sliding window of
5000 nucleotides (you can use the letterFrequencyInSlidingView
function) and visualise it (see an example below). On that plot, show
the mean GC content and highlight the regions that have a GC content >
0.5 and < 0.35.
Figure 4.1: GC content for chomosome 2L (10 - 11 Mb)
► Question
Visit the UniProt web page at https://www.uniprot.org/ and download the manually reviewed complete human proteome (containing about 20500 proteins). To do so, choose Proteomes on the main page, then search for Homo sapiens, select it among the list of hits, choose the reviewed Swiss-Prot release, and download all entries in compressed fasta format.
Alternatively, under Proteins UniProt Knowledge base, choose Reviewed (SwissProt), then in the left panel, select Human (20,420), then Download, choosing FASTA (canonical) and compressed format (video).
► Question
Load the file into R. Beware though that these are protein sequences,
composed of amino acids. Read the ?readDNAStringSet manual page to
find a more appropriate function. How many sequences are there? What’s
the average. max and min length of the proteins?
► Question
Load the up_selected data from the rWSBIM1322 data (version 0.1.1
or later) and use it to create a subset of the object created
above. To to so, you will need to match the protein identifiers from
that vector and the names of the AAStringSet above.
► Question
Among the proteins of interest (that where also found in the original
data), how many match the following patterns: DDVF, DEVF EDVF or
EEVF? Do you consider this to be a high number?
The identification of patterns in sequences (not specifically
biological sequences) is called pattern matching, which defines a
whole syntax to define patterns. In particular, it is possible to
define letter D or E as [DE]. To find the pattern of interest, use
the str_detect function from the stringr package.
► Question
To assess if there is an overrepresentation of that pattern in the sequences of interest, repeat the calculation on all the other proteins.
Page built: 2026-03-31 using R version 4.5.0 (2025-04-11)